Hydrogen
Hydrogen, the most abundant element in the Universe, has a gravimetric energy density three times that of natural gas and is currently being used to power fuel cell vehicles in California. We study materials that can be used in next-generation hydrogen fuel cells to generate electricity more safely and efficiently.
Hydrogen strongly affects the electronic and structural properties of many materials. It can bind to defects or to other impurities, often eliminating their electrical activity: this effect of defect passivation is crucial to the performance of many photovoltaic and electronic devices. A full understanding of hydrogen in solids is required to support development of improved hydrogen-storage systems, proton-exchange membranes for fuel cells, and high-permittivity dielectrics for integrated circuits. In chemistry and in biological systems, there have also been many efforts to correlate proton affinity and deprotonation with host properties. We have performed systematic studies, based on first-principles methods, of hydrogen in a wide range of hosts.
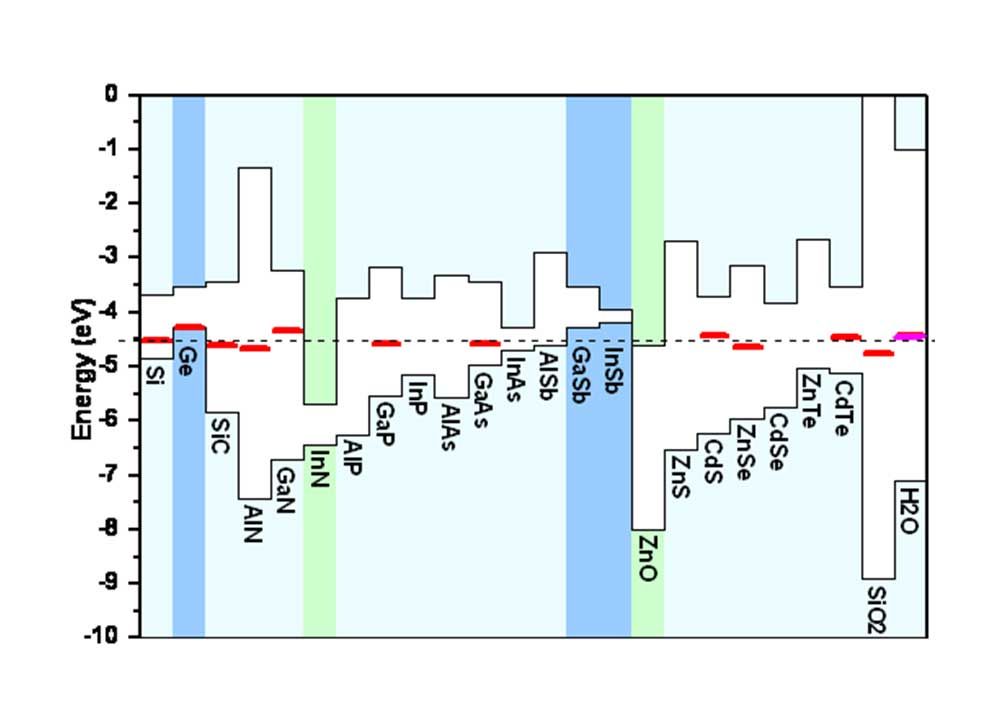
Universal alignment of hydrogen levels
Our systematic first-principles studies for hydrogen in a wide range of hosts revealed the existence of a universal alignment for the electronic transition level of hydrogen in semiconductors, insulators and even aqueous solutions. This alignment allows the prediction of the electrical activity of hydrogen in any host material once some basic information about the band structure of that host is known. A physical explanation was presented that connects the behavior of hydrogen to the lineup of electronic band structures at heterojunctions. The universal alignment level is at about -4.5 eV below the vacuum level. [1]
Hydrogen storage
Using hydrogen as an energy carrier will potentially allow us to reduce our dependency on fossil fuels and to make the transition to a greenhouse gas neutral society. A major challenge involves the efficient generation of hydrogen from renewable sources. Solar energy stands out as a source with high potential, which could be used to generate hydrogen in a photoelectrochemical cell.
Another major challenge is, that appropriate materials for storing hydrogen are still missing, in particular for mobile applications. In the recent years a variety of different material classes has been proposed as potential candidates, including metal hydrides, carbon based nanostructures, metal-organic frameworks and doped polymers, to mention only some of them. First-principles simulations allow to systematically investigate the microscopic mechanisms which drive the hydrogen sorption characteristics in these materials. We employ density functional theory to investigate the mechanisms of point-defect kinetics and their relevance for the hydrogen uptake and release in systems like in high-capacity hydrogen storage materials such as MgH2, AlH3, NaAlH4 , LiBH4 and Li4BN3H10. [1] The goal of these studies is to provide interpretations for experimental observations and also to contribute to the design of better storage materials, i.e. to propose target-oriented engineering strategies to improve the control of the hydrogen sorption kinetics.
Our goal is to elucidate the physics and chemistry of hydrogen interactions with hydrogen storage materials by performing first-principles calculations based on density functional theory (DFT). By specifically addressing the behavior of hydrogen-related and other point defects, we are able to investigate the kinetics of hydrogen uptake and release, the decomposition of complex hydrides, and the effect of adding impurities to the system. [2,3] Our approach takes into account that defects and impurities in non-metallic systems can occur in charge states other than the neutral state; this important aspect of the problem had not been addressed in previous computational studies performed to date, and our investigations show that it has extremely important consequences for defect concentration and diffusion. Other groups have adopted our methodology.
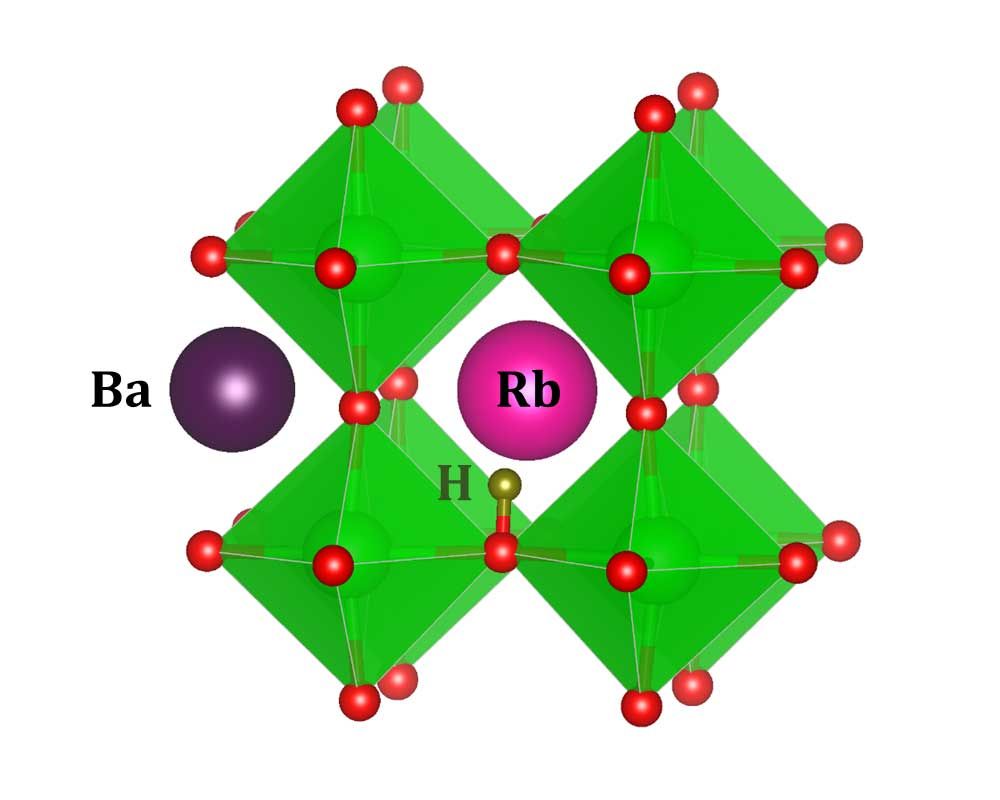
Hydrogen transport
Solid-state hydrogen fuel cells require crystalline electrolytes with fast kinetics for hydrogen ionic conduction. Uniquely among the elements on the periodic table, hydrogen can favorably ionize as a positively charged proton or as a negatively charged hydride ion. Typical proton conductors incorporate hydrogen as an extrinsic impurity (Hi+), and migration proceeds by protons rotating around and hopping between native anions (usually oxygen, e.g. in BaZrO3). Hydride conductors like BaH2, on the other hand, contain hydrogen intrinsically, and conduction proceeds through a vacancy-mediated mechanism. These vacancies are donor defects (VH+). Certain novel crystals, called oxyhydrides, in which both oxygen and hydrogen occupy anionic sites in the lattice, have characteristics of both of these kinds of systems and have been demonstrated to be among the best solid-state hydrogen (specifically, hydride) conductors.
Using our formalism, we investigate strategies to promote defects favorable to conduction in these materials. We propose different synthesis conditions and prospective acceptor dopants to encourage the formation of defects necessary for conduction. We also identify defects that may hinder conductivity, for instance, by destabilizing the materials, and offer engineering solutions to circumvent their detrimental effects. [1,2]
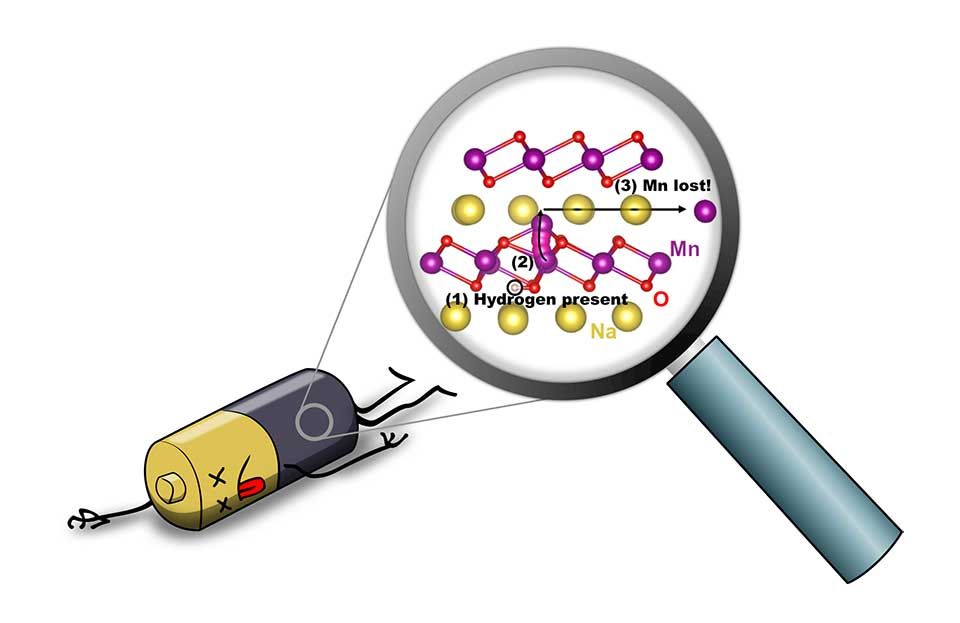
Hydrogen as a cause of degradation in battery materials
Sodium-ion batteries are being researched intensely as an alternative to lithium-ion batteries. Their performance is good, and sodium, an alkali metal closely related to lithium, is cheap and abundant. The challenge is that sodium-ion batteries have shorter lifetimes than their lithium-based siblings.
We have uncovered a reason for the loss of capacity that occurs over time in sodium batteries: the unintended presence of hydrogen, which leads to degradation of the battery electrode. Hydrogen is commonly present during the fabrication of the NaMnO2 cathode material, or it can be incorporated from the environment or from the electrolyte. Hydrogen is known to strongly affect the properties of electronic materials. We found that hydrogen can very easily penetrate the material, and that its presence enables the manganese atoms to break loose from the manganese-oxide backbone that holds the material together. This removal of manganese is irreversible and leads to a decrease in capacity and, ultimately, degradation of the battery.
Earlier research had shown that loss of manganese could take place at the interface with the electrolyte or could be associated with a phase transition, but it did not really identify a trigger. Our results show that the loss of manganese can occur anywhere in the material, if hydrogen is present. Because hydrogen atoms are so small and reactive, hydrogen is a common contaminant in materials. Now that its detrimental impact has been flagged, measures can be taken during fabrication and encapsulation of the batteries to suppress incorporation of hydrogen, which should lead to better performance. [1]