Complex Oxides
Interfaces between certain complex oxides have been shown to spontaneously form a two-dimensional electron gas (2DEG) with a carrier density corresponding to 1/2 electron per interface unit cell - one observes metallic behavior at the interface between two insulators. We have been working on understanding these interfaces: Where do the carriers come from, into which material do they go, how can surface terminations play a role, and why are some interfaces insulating even though they have the carrier density corresponding to a 2DEG? We have also investigated the bulk materials themselves, without the presence of an interface. Many of the materials are perovskite transition-metal oxides that are largely unexplored in the literature. We have also been investigating stannates and germanates. Our efforts have focused on the mechanisms determining carrier mobility, band gaps, charge localization, and the behavior of defects.
Read our popular-science account of this research topic at FunSizePhysics.com.
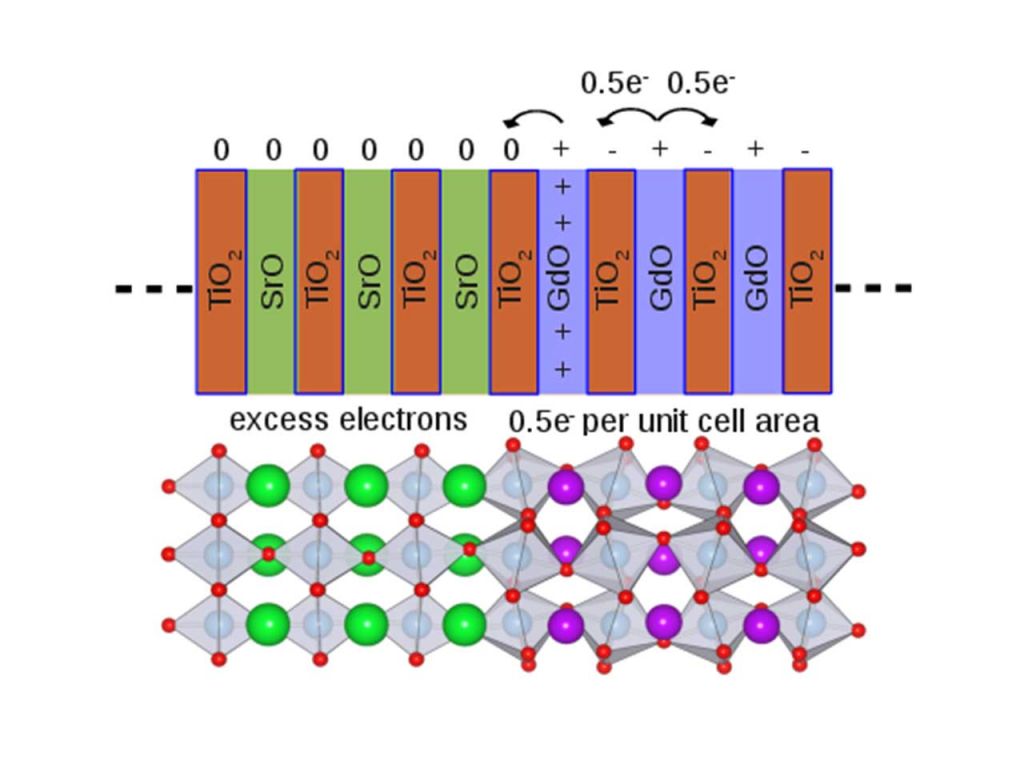
Two-dimensional electron gas at complex oxide interfaces
When an interface is formed between a polar and a nonpolar material, the layer in the polar material adjacent to the nonpolar material donates 1/2 electron to the right, i.e., to a "bulk-like" layer in the polar material. It is also trying to donate 1/2 electron to the layer to its left—but that layer belongs to a nonpolar material and does not need that electron to satisfy its bonding. This electron is therefore in principle available as a free electron, and the atomic layer on the polar side of the interface effectively acts as a layer of donors with a density of 1/2 of the areal density of atoms at the interface. In most materials, this is on the order of a few times 1014 cm-2; i.e. a huge density compared to what is typically achieved in two-dimensional electron gases (2DEGs) at conventional interfaces.
The polar discontinuity at the SrTiO3/LaAlO3 interface (STO/LAO) can therefore in principle sustain an electron density of 3.3×1014 cm−2 (0.5 electrons per unit cell). However, experimentally observed densities are more than an order of magnitude lower. Using a combination of first-principles and Schrödinger-Poisson simulations we have shown that the problem lies in the asymmetric nature of the structure, i.e., the inability to form a second LAO/STO interface that is a mirror image of the first, or to fully passivate the LAO surface. Our insights apply to oxide interfaces in general, explaining for instance why the SrTiO3/GdTiO3 interface has been found to exhibit the full density of 3.3×1014 cm−2. [1]
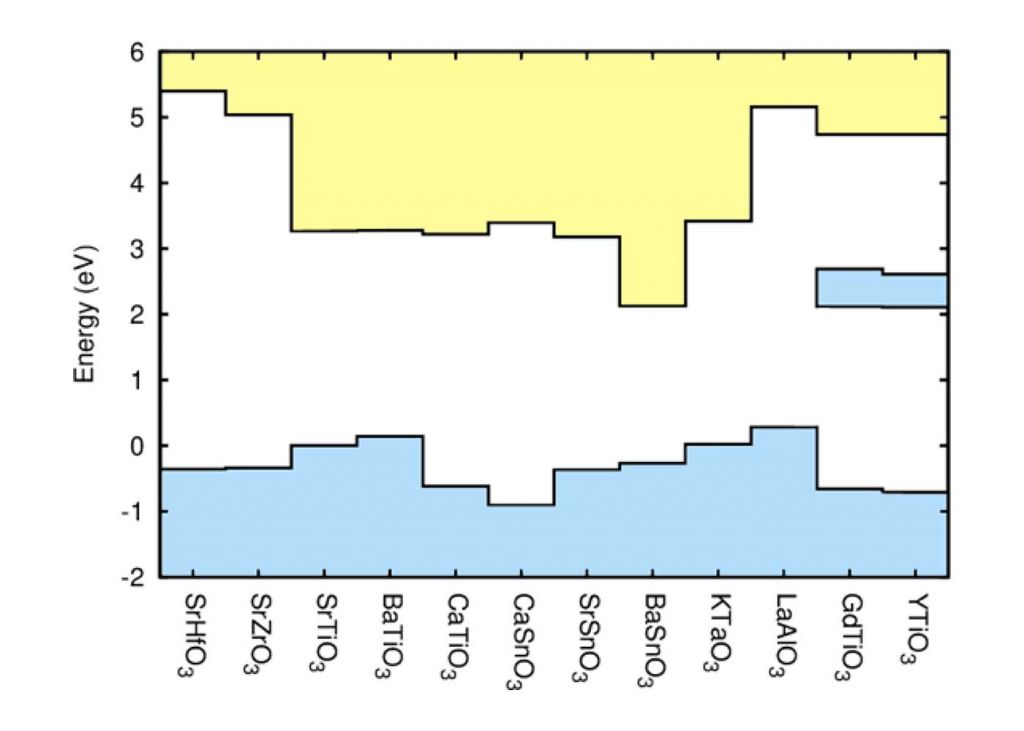
Band alignment at complex oxide interfaces
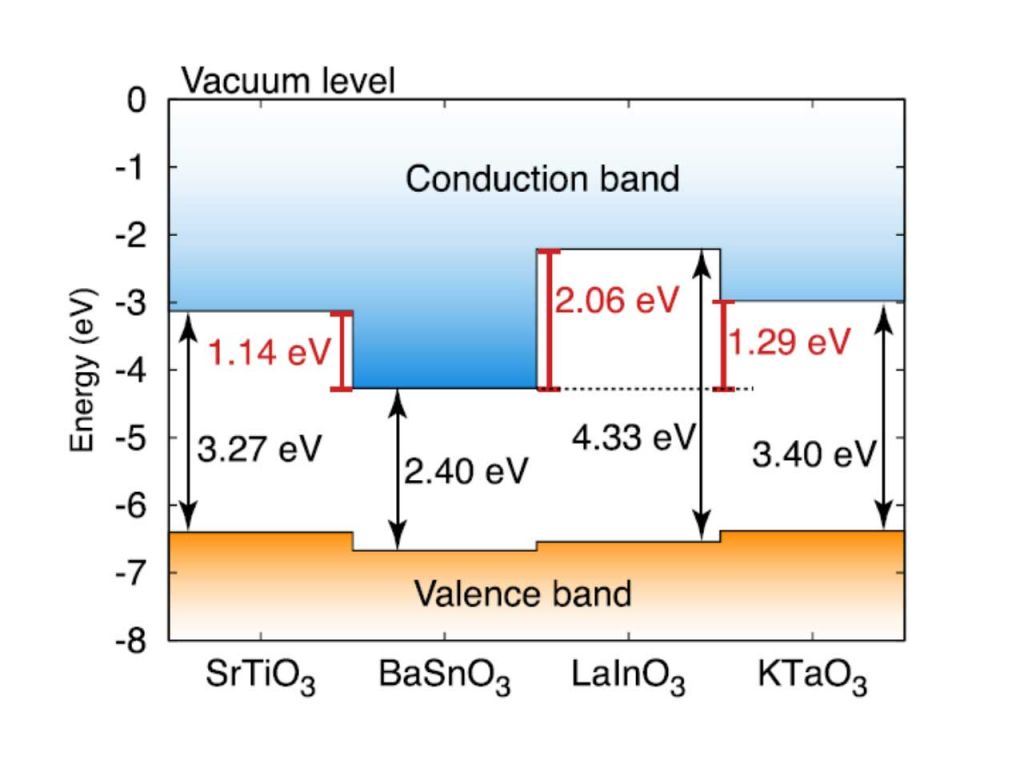
For these interfaces, a key issue is the band alignment, which determines on which side of the interface the 2DEG will reside, as well as the degree of confinement. We have use hybrid density functional calculations to determine the band alignments of a number of complex oxides, considering materials with different types of conduction-band character, polar or nonpolar character and band insulators as well as Mott insulators. We suggest promising materials combinations that could lead to a 2DEG with optimized properties, such as high density and high mobility – pointing to BaSnO3 as an interesting candidate as a host material for a 2DEG given its high room temperature mobility; much higher than that of SrTiO3. [1]
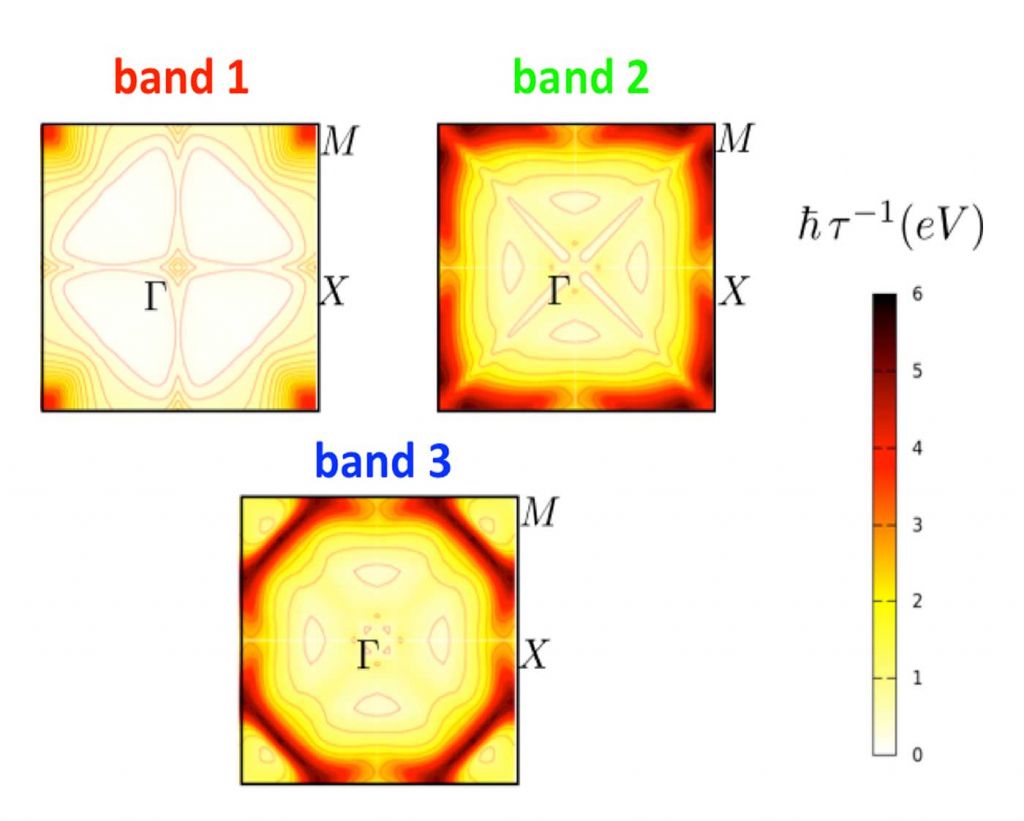
Understanding the mobility of complex oxides
The mobility of the electrons in the 2DEG is critical to potential applications, and we have therefore explored the mechanisms that govern the mobility.
Strain can affect the energetic ordering and effective mass of the lowest conduction-band states in SrTiO3, made up of Ti 3d states. We have predicted that biaxial stress in the (001) or (110) planes results in the lowest-energy conduction-band state having significantly smaller electron mass in the in-plane directions compared to the unstrained SrTiO3, thus suggesting that pseudomorphic growth is a promising route to increasing the electron mobility in epitaxial films. [1]
We have also investigated the electronic and vibrational spectra of SrTiO3, as well as the coupling between them. We compute electron-phonon scattering rates for the three lowest-energy conduction bands and use Boltzmann transport theory to calculate the room-temperature mobility of SrTiO3. The results agree with experiment and highlight the strong impact of longitudinal optical phonon scattering. Our analysis provides important insights into the key factors that determine room-temperature mobility, such as the number of conduction bands and the nature and frequencies of longitudinal phonons. Such insights provide routes to engineering materials with enhanced mobilities. [2]
Metal-insulator transition in ultra-thin SrTiO3 layers
In addition to the metallic behavior seen at the interface between complex oxides such as SrTiO3 and GdTiO3, experimental results for ultrathin SrTiO3 layers inserted in GdTiO3 reveal a transition from metallic to insulating behavior, and suggest a strong interplay between electron-electron interaction and lattice distortions. [1] We have shown that a metal-to-insulator transition can occur in SrTiO3 at extreme doping levels. We find that doping with 1/4 electron per Ti atom produces a metallic phase as expected, but that adding 1/2 electron per Ti results in a charge-ordered Mott-insulating phase. These excess electrons occupy the otherwise empty Ti 3d bands. This Mott-insulator phase was also found to occur in calculations of SrTiO3/LaAlO3 and SrTiO3/GdTiO3 heterostructures with ultrathin SrTiO3 layers. [2]
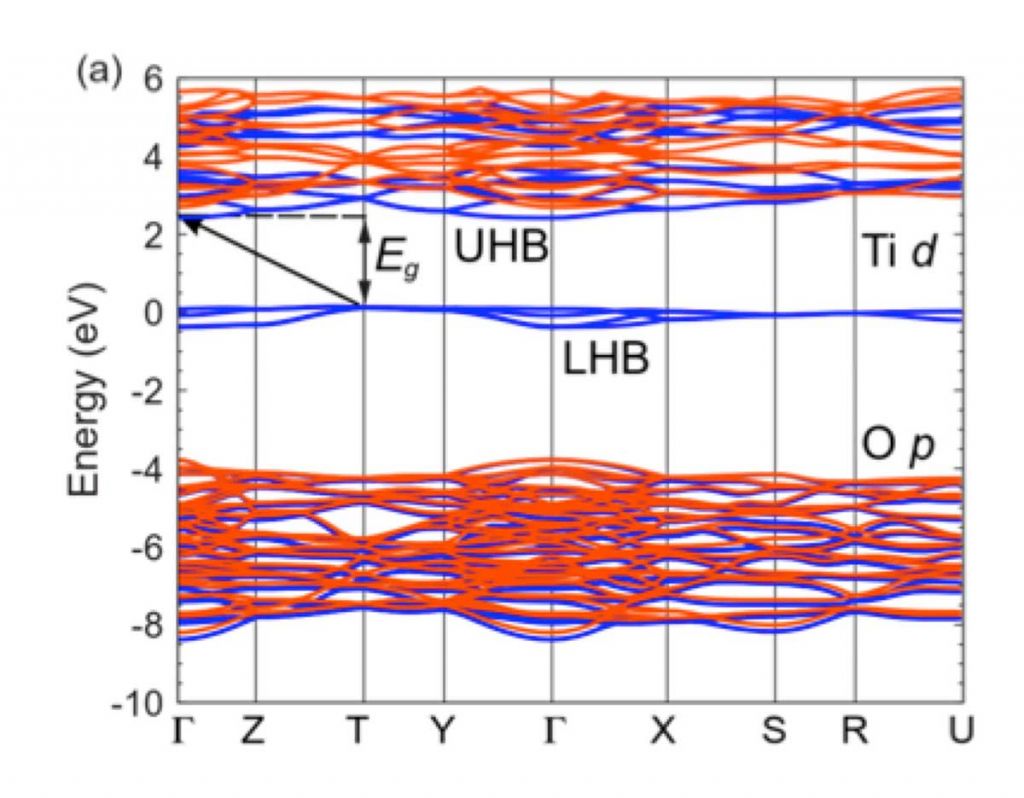
The Mott-Hubbard gap of the rare-earth titanates
YTiO3 is a representative member of the rare-earth titanate Mott insulators. These materials have become the focus of great interest because of their use in complex-oxide heterostructures, and because of the potential for switching behavior to be exploited in novel electronic devices. We are scrutinizing the fundamental properties, such as the magnitude of the Mott-Hubbard gap. Commonly accepted values for the gap in YTiO3 and other rare-earth titanates are in the range of 0.2 – 0.7 eV, based on optical conductivity spectra. However, we find a 2 eV gap in our hybrid functional calculations. [1] This has been confirmed by luminescence experiments.
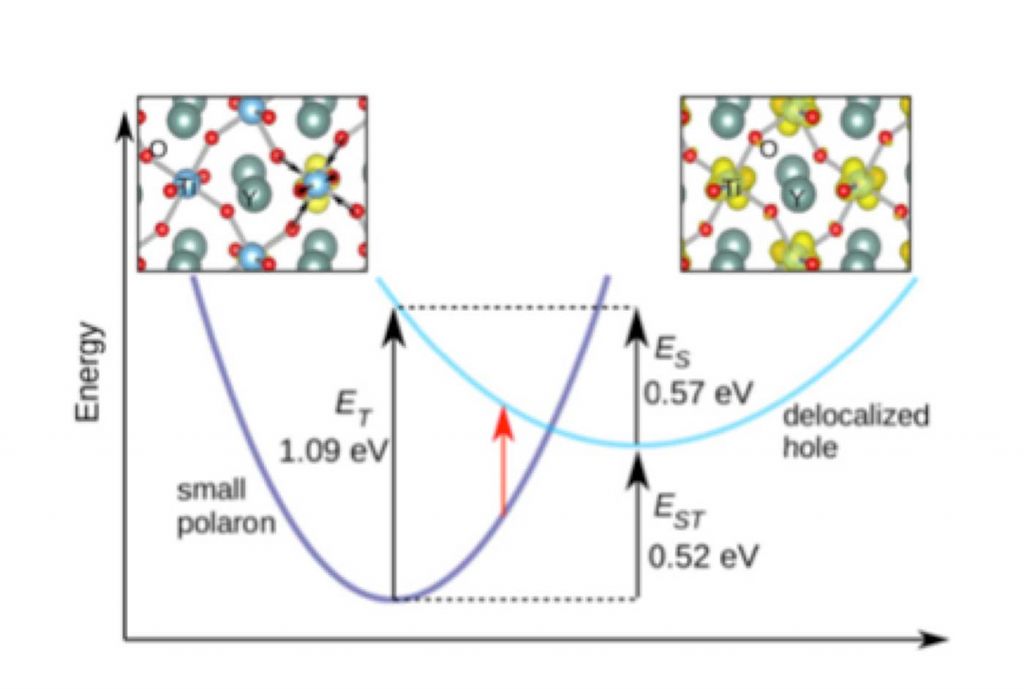
Small polarons in rare-earth titanates
We have proposed that the optical conductivity onset at 0.7 eV for YTiO3 is caused by the absorption of an electron in the lower Hubbard band to a small hole polaron state; holes tend to become self-trapped on the Ti lattice sites. The presence of small hole polarons is consistent with experimental reports of the rare-earth titanates being p-type.
Since the magnitude of the Mott-Hubbard gap reflects the strength of the intra-atomic Coulomb repulsions in the material, knowing its value is essential for a correct understanding of the correlated nature of d-orbital derived bands. Our work can also serve as a guide to further developments in the theory of Mott insulators.
Small polarons also dominated the defect physics in these materials. [1,2]
High-mobility oxides
SrTiO3 may be the most widely studied perovskite oxide, but its application in electronic devices is limited by its low room-temperature mobility. Therefore the search is on for materials that exhibit higher mobility. We identified WO3 as a potential candidate. [1] Looking beyond transition-metal oxides, moving to materials in which the conduction band is composed of s states rather than d states is attractive. This has drawn attention to the stannates. BaSnO3 (BSO) has become a widely studied material thanks to its demonstration of a record room-temperature electron mobility (320 cm2/Vs) among wide-band-gap oxides. It has been proposed in applications as a transparent conducting oxide. SrSnO3 (SSO) has also been studied for various electronic applications. Much of the research into BSO has focused on taking full advantage of its high mobility through electron doping (often with La as a donor dopant) and band engineering.
We have used first-principles methods to rigorously calculate the mobility in BSO, accounting for both LO-phonon scattering and ionized impurity scattering. [2] The latter of these two processes particularly limits the highest achievable mobility, but it can be avoided largely through modulation doping or polar-discontinuity doping. Such doping techniques can readily be achieved in BSO on account of its large conduction band offset compared with the closest lattice-matched barrier materials. [3]
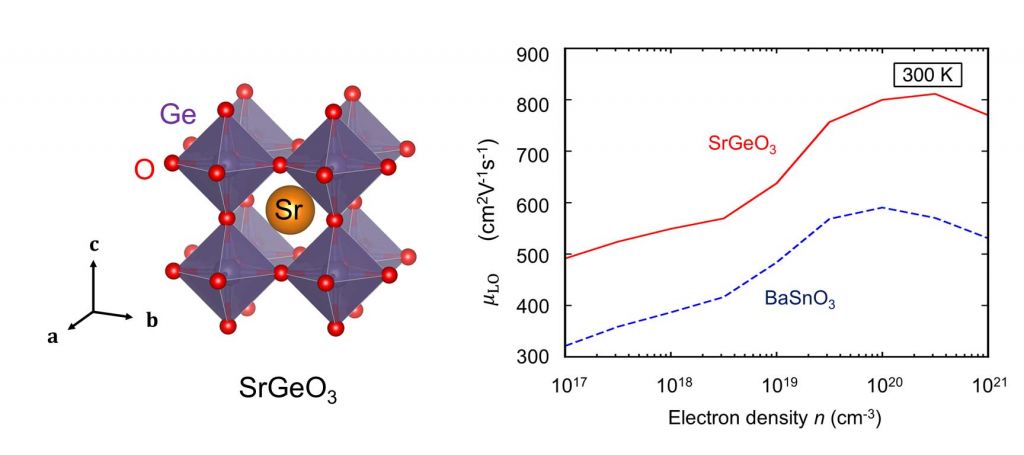
Germanates
Perovskite oxides with germanium on the B-site have attracted little research attention in comparison to those containing tin, such as BaSnO3. The cubic germanates, specifically SrGeO3 and BaGeO3, share many important characteristics with BaSnO3, including highly dispersive lowest conduction bands, good lattice matching with common substrate materials such as LaAlO3 and SrTiO3, and wide band gaps suitable for applications in optoelectronics . Using first principles calculations, we find the germanates to possess small carrier effective masses, both for electrons and holes, rendering them attractive as n- or p-type conductors. We also calculate electron mobility in SrGeO3, which we find to be 50% larger than that of BaSnO3. Our results should encourage experimental development of the germanates for transparent electronics. [1]